






Frequently Asked Questions
The main purpose of SwissSimilarity is to provide user-friendly interface to perform ligand-based virtual screening of chemical libraries. By employing different molecular fingerprints, SwissSimilarity is able to retrieve compounds that are similar to a user’s query molecule. According to the similarity principle, the output molecules provided by SwissSimilarity are expected to be enriched in compounds targeting the same proteins as the query molecule. Thus, the website can be used by drug discovery scientists in their hit finding and scaffold hopping activities as well as in structure-activity relationships studies.
Wide range of compound libraries are available for screening using SwissSimilarity, all of them are divided in four groups: 1. Drugs and clinical candidates; 2. Bioactive compounds; 3. Commercially available compounds; 4. Synthesizable molecules.
The “Drugs” group consists of the following compound collections:
The “Bioactive” group includes the following chemical libraries:
The “Commercial” group consists of the following chemical libraries:
The “Synthesizable” group includes libraries of compounds that can be readily synthesized and delivered:
Available methods are based on usage of various molecular fingerprints/vectors, that encode either 2D molecular structure or 3D conformations of a compound.
Among 2D methods, there are path-based FP2 fingerprint, extended-connectivity fingerprint with diameter 4 (ECFP4), MinHash fingerprints (MHFP6, up to 6 bonds), 2D pharmacophore fingerprints and extended reduced graph fingerprints (ErG). Additionally, we have prepared ECFP4 fingerprints based on scaffold and generic scaffold (in which all atoms are converted to carbons and all bonds are converted to single bonds) of each library compound, thus allowing to screen for molecules having the same scaffold as the query molecule. Last but not least, two non-superpositional 3D methods are available: Electroshape 5D vectors (ES5D) and extended 3D fingerprints (E3FP).
As a rule of thumb, FP2, ECFP4, MHFP6, and to some extent scaffold-based ECFP4, can be useful for finding structures that are similar to the query compound (e.g. for structure-activity relationship studies) because those fingerprints describe a molecule in high level of detail and they focus on detecting similar substructures in different compounds. On the other hand, 2D-pharmacophore, ErG, ES5D as well as generic scaffold-based ECFP4 fingerprints may be more useful for hit finding and scaffold hopping activities. Pharmacophore and ErG focus on detecting similar arrangement of pharmacophoric points (hydrogen donors/acceptors, basic/acidic/hydrophobic moieties, etc) in compounds, while ES5D concentrates on finding compounds with similar shape as well as similar projection of lipophilicity and electrostatics in the 3D space.
More about the implemented fingerprinting methods you can find in the respective publications:
ECFP4: Rogers D. & Hahn M., “Extended-Connectivity Fingerprints”, J Chem Inf Model (2010);
MHFP6: Probst D. & Reymond J.L., “A probabilistic molecular fingerprint for big data settings”, J Chemoinformatics (2018);
ErG: Stiefl N. et al., “ErG: 2D pharmacophore descriptions for scaffold hopping”, J Chem Inf Model (2006);
2D-pharmacophore: Gobbi A. & Poppinger D., “Genetic optimization of combinatorial libraries”, Biotech and Bioeng (1998), https://www.rdkit.org/docs/RDKit_Book.html#representation-of-pharmacophore-fingerprints;
E3FP: Axen S.D. et al., “A simple representation of three-dimensional molecular structure”, J Med Chem (2017);
ES5D: Armstrong M.S. et al., “ElectroShape: fast molecular similarity calculations incorporating shape, chirality
and electrostatics”, J Comput Aided Mol Des (2010);
Combined score is calculated from similarity values obtained using two different molecular representations: FP2 fingerprints (2D method) and ES5D vector (3D method). Mathematically, the combined score is a probability that a given couple of molecules share a common protein target. It was obtained using logistic regression with two features:
f(FP2,ES5D)=(1+exp(-a0-a1FP2-a2ES5D))-1, where:
• FP2 and ES5D are similarities between FP2 fingerprints and ES5D vectors, respectively;
• a0, a1 and a2 are parameters learned by the model to predict possible protein targets for a small molecule based on molecular similarity to known bioactive compounds.
f(FP2,ES5D) ranges from 0 for totally dissimilar molecules to 1 for perfectly identical molecules. This combined score was found to perform significantly better for drug-like molecules than the similarity assessed by FP2 or Electroshape-5D separately.
First of all, chemical libraries available in SwissSimilarity were cleared from compounds with less than six heavy atoms or with molecular weight higher than 1500 g/mol or containing unusual atoms (other than H, C, N, O, S, P, B, F, Cl, Br and I) in its largest fragment. As the second preparatory step, compounds underwent standardization, which included keeping only the largest fragment as well as dearomatization, neutralization and dehydrogenization. Afterwards, the most frequent tautomeric state was calculated for each structure and obtained dominant tautomers were further used to prepare all 2D fingerprints used in SwissSimilarity: FP2, ECFP4, MHFP6, pharmacophore and ErG. For 3D methods, the major protonation state (pH 7.4) was additionally calculated and further used to generate conformers, based on which E3FP and ES5D fingerprints were prepared.
All submitted query structures undergo standardization. In particular, only the largest fragment of the query molecule is left, and it is further converted to Kekule form (dearomatized), neutralized and dehydrogenized. If a user opts for a 3D-based method, then major protonation state at pH 7.4 of the molecule is additionally calculated and further used for 3D conformers generation. It is important to highlight that the most frequent tautomeric state is not calculated for the input compounds, while this has been done for all compounds from the chemical libraries. Thus, a user has the possibility to search for compounds similar to a molecule that is in its less frequent tautomeric state.
Both forms are acceptable since all query compounds undergo standardization, which includes dearomatization. Thus, all compounds submitted in aromatic form will be converted to Kekule one.
Only 3D-based methods (ES5D, E3FP and Combined (FP2+ES5D)) account for conformational flexibility of small molecules. ES5D-based screening relies on the comparison of 20 conformers of the query molecule with 20 conformers of each molecule in a chosen library. E3FP-based screening compares top 3 low energy conformers of the query compound with top 3 low energy conformers of library compounds.
Time required to complete the screening strongly depends on chosen library/method combination. Approximate time to run the screening appears on the submission page in the upper-left corner of the table when user moves the cursor from one method/library combination to another. However, this time does not consider waiting time in the queue that depends on machine’s load at a given time point. Position of your job in the queue is displayed after job’s submission. With the fixed library size, the speeds of calculation using different methods rank this way : ECFP4 < FP2 < Scaffold < Generic Scaffold < ES5D < Combined < Pharmacophore < ErG < E3FP < MHFP6. As an example, screening of 2,059,285 molecules of the CHEMBL29 database using ECFP4 fingerprints takes about 9 sec, while screening the same library with MHFP6 fingerprint takes 180 seconds.
Preparation of the E3FP and ES5D libraries requires significant computational resources because of the 3D conformers generation step. Thus, it has been decided to make those screening methods available only for libraries belonging to the groups “Drugs” and “Bioactive compounds” as well as ZINC libraries.
All similarity scores obtained with different methods range from 0 for totally dissimilar compound to 1 for identical ones. However, for a given pair of compounds, similarity values can vary significantly depending on the fingerprinting method chosen for molecular representation. To better understand and compare similarity values obtained using different methods, we estimated the probability that two bioactive compounds have a protein target in common as a function of their similarity calculated with different methods. To estimate those probabilities, we used a dataset containing 10 million pairs of compounds sharing the same protein target and 100 million pairs of randomly selected compounds. Obtained probability curves are shown here:
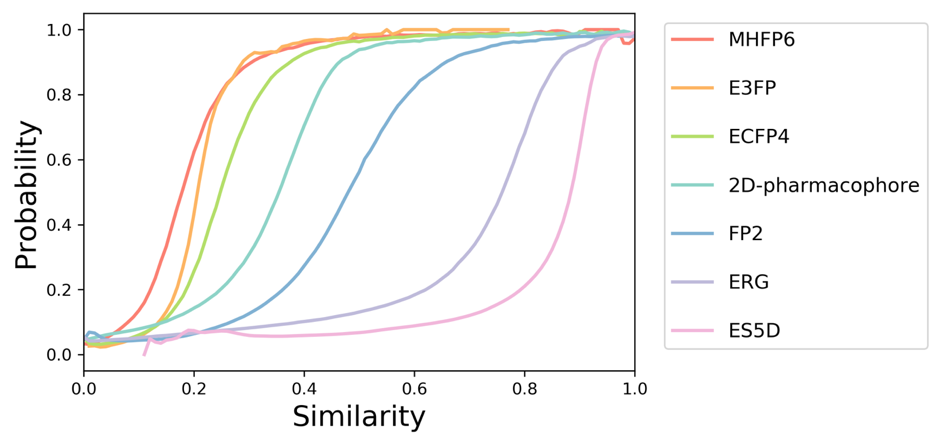
As an example, the curves indicate that a compound that has been retrieved with a MHFP6-based similarity of 0.2 to a query molecule has the same probability to share a common protein target with it as another compound that has FP2-based similarity of 0.5 with a query molecule.
The website is optimized for Google Chrome or Mozilla Firefox. Other browsers can work but without any guarantee. Also, ensure that your network/infrastructure allows access to Web services and that pop-up windows are permitted.